
Artículo
Hipercolesterolemia Severa: detección y descripción Clínica, Bioquímica y Genética en el Municipio de General Pueyrredón. Premio “Arturo Alió” – Clínica Médica y Especialidades Médicas, edición 2022
Resumen
La hipercolesterolemia severa (HS) se define cuando el valor del Colesterol-LDL (C-LDL) es mayor a 190 mg/dL. Por otro lado la hipercolesterolemia familiar (HF) es el desorden monogénico autosómico dominante más común, que causa aterosclerosis prematura severa y elevada mortalidad en los individuos afectados sin un tratamiento temprano; está determinada principalmente por la existencia de variantes en genes críticos que intervienen en el metabolismo de las lipoprteínas de baja densidad (LDL). El objetivo del presente trabajo consiste en realizar un análisis detallado clínico, bioquímico y genético al subgrupo con HS en el Municipio de General Pueyrredón Pueyrredón y detectar a los portadores de HF. Se describen estas tres características -clínicas, bioquímicas y genéticas- con el fin de demostrar la relación existente entre el fenotipo con elevación extrema de C-LDL y la presencia de variantes genéticas de esta población.
Abstract
Severe hypercholesterolemia (HS) is defined when the value of LDL-Cholesterol (LDL-C) is greater than 190 mg/dL. On the other hand, familial hypercholesterolemia (FH) is the most common autosomal dominant monogenic disorder, which causes severe premature atherosclerosis and high mortality in affected individuals without early treatment; It is mainly determined by the existence of variants in critical genes involved in the metabolism of low-density lipoproteins (LDL). The objective of this work is to carry out a detailed clinical, biochemical and genetic analysis of the subgroup with HS in the Municipality of General Pueyrredón Pueyrredón and to detect FH carriers. These three characteristics -clinical, biochemical and genetic- are described in order to demonstrate the relationship between the phenotype with extreme elevation of C-LDL and the presence of genetic variants in this population.
Palabras clave: Hipercolesterolemia severa, Hipercolesterolemia familiar, Aterosclerosis, Colesterol,Lipoproteínas baja densidad.
Keywords: Severe hypercholesterolemia, Familial hypercholesterolemia, Atherosclerosis, Cholesterol, Low-density lipoproteins.
Fecha de recepción: 09/08/2022
Fecha de aceptación:09/03/2023
INTRODUCCIÓN
En 2016, la Sociedad Internacional de Aterosclerosis respaldó la declaración publicada por varios expertos comprometidos con la definición de las hipercolesterolemias severas, estableciendo que el riesgo de enfermedad cardiovascular aterosclerótica (ECV) se relaciona directamente con la exposición crónica y acumulativa de los niveles elevados de las lipoproteínas de baja densidad (LDL) (1). El rol de las LDL como causante de ECV se ha ratificado globalmente, exponiendo evidencias epidemiológicas, genéticas, clínicas de intervención y difundiendo la concientización de su importancia (2).
La hipercolesterolemia severa (HS) se define cuando el valor del Colesterol-LDL (C-LDL) es mayor a 190 mg/dL. Las HS pueden deberse de forma secundaria a otras patologías como la colestasis, el hipotiroidismo o la enfermedad renal, pero por otro lado pueden deberse a causas genéticas -monogénicas o poligénicas-, epigenéticas o ambientales y a la vez presentarse de manera combinada.
La hipercolesterolemia familiar (HF) es el desorden monogénico autosómico dominante más común, que causa aterosclerosis prematura severa y elevada mortalidad en los individuos afectados que no tuvieron oportunidad de tratamiento temprano. Según cifras clásicas, se estimaba que 1/500 sujetos eran portadores heterocigotas de HF (HFHe) y 1/1.000.000 serían homocigotas (HFHo) manifestando las formas más severas de la enfermedad (3). En los últimos años, se ha generado una reactivación en el estudio de HF, especialmente en países como Holanda, Inglaterra, Dinamarca, España y Australia, que han instrumentado registros poblacionales revelando prevalencias significativamente más altas que rondan 1/200-1/300 para heterocigotas (4,5). En nuestro país se ha llevado a cabo el Primer Programa de Detección de HF (Estudio Da Vinci) que se lanzó en el Distrito de Gral Pueyrredón, avalado por las Universidades FASTA y Universidad de Buenas Aires, y permitió conocer el primer dato de prevalencia de HFHe obtenido en nuestro país de 1/295, que fue publicado en el año 2018 (6).
La importancia de conocer la prevalencia local de HF y la detección temprana de los casos índices y sus familiares se basa en la premisa de que la HF está subdiagnosticada en el mundo y por ende subtratada, teniendo en cuenta que en los últimos años los avances en el desarrollo de moléculas novedosas dieron lugar a nuevas alternativas de tratamiento que ofrecerían mejor pronóstico a los pacientes con HF (7). Por lo tanto, se asume que las HS, entre ellas la HF, son una indicación directa de tratamiento con estatinas, y combinaciones de fármacos hipolipemiantes emergentes.
La HF está determinada principalmente por la existencia de variantes en genes críticos que intervienen en el metabolismo de las LDL. En circunstancias normales las partículas de LDL son captadas por los tejidos hepático y extrahepáticos, a través de receptores específicos (rLDL) ubicados en la membrana de las células, que reconocen a la apolipoproteína B (apoB) de las LDL. En el citoplasma de las células, la lipoproteínas se degradan, y su colesterol liberado regula la biosíntesis del colesterol intracelular y la producción de más receptores que permitirán la captura de más partículas de LDL circulante, manteniendo los niveles plasmáticos adecuados. La funcionalidad de los rLDL es fundamental para que este mecanismo funcione correctamente y la pro-proteína convertasa subtilisina kexina tipo 9, conocida como PCSK9, ejerce un rol relevante en reciclar a los rLDL de membrana, con lo cual se infiere que, si esta proteína es muy activa, los rLDL se degradan a una tasa mayor a la esperada, impactando en los niveles de LDL circulante. Asimismo, siendo la apoB de la LDL el ligando específico del rLDL, alteraciones genéticas en su estructura afectarán el reconocimiento de las LDL circulantes (8).
Las variantes genéticas en el gen del receptor del LDL (R-LDL) que conducen a la pérdida de función del rLDL, ocurren en el 90% de los casos, menos común son las mutaciones en el gen de la apoB (APOB) que reduce la unión de la apoB de la LDL con el receptor - 5% de las variantes- y aún menos frecuentes -alrededor del 2%- se atribuyen a mutaciones con ganancia de función en el gen PCSK9, que codifica la proteína encargada de la degradación del rLDL (9). Asimismo, la forma autosómica recesiva de la HF se asocia a mutaciones en el gen LDLRAP1, que codifica una proteína adaptadora del receptor LDL, interfiriendo en la captación de las LDL; y otro gen relevante que suele explicar casos de HF es APOE, ya que la apoproteína E también es ligando del rLDL y las isoformas homocigota E4/E4 se asocian a HS.
Por lo tanto, variantes patogénicas en estos genes mencionados afectan el catabolismo de las LDL e inducen la elevación de C-LDL plasmático.
En relación con el diagnóstico de HS, el valor de corte de C-LDL>190 mg/dL debe advertir la posible presencia de HF, lo cual sumado a datos de historia familiar o personal de ECV y/o hipercolesterolemia, presencia de xantomas tendinosos y/o arco corneal en jóvenes, incrementan la certeza del diagnóstico. Para el diagnóstico clínico de HF se suelen usar puntajes o scores como el propuesto por las Clínicas Holandesas de Lípidos (DUTCH), la cual incluye los signos y antecedentes mencionados (10). Si bien se valora el estudio genético de las variantes patogénicas en la medicina personalizada, varios autores acuerdan que la investigación de la variante genética causal no sería esencial para el diagnóstico y decisión de tratamiento, los cuales se basarían en el nivel de C-LDL y los antecedentes personales y/o familiares de cada paciente (1). Desde el punto de vista de la Salud Pública, estas recomendaciones serían importantes, sin embargo, hay evidencias que desafían estos conceptos, demostrando una destacada superposición de niveles de C-LDL entre portadores de HF no tratados y no portadores (11), y la evaluación prospectiva ha revelado que en cada rango de concentración de C-LDL, los pacientes con variantes en los genes relacionados con HF presentan sustancial incremento de ECV en comparación a los no portadores (11).
Cabe destacar que los estudios genéticos, aún realizados por secuenciación de última generación (NGS) demuestran que entre los individuos que presentaron un diagnóstico clínico-bioquímico certero, entre el 20 al 40% de los casos son negativos para el hallazgo de una variante patogénica de su fenotipo. (12, 13). Los casos con claro fenotipo de HF y sin variante presente en los genes clásicos, se atribuyen en su mayor parte a causas poligénicas asociadas al incremento de C-LDL. Destacados grupos de investigadores proponen la determinación del Score de Riesgo Genético (GRS) para explicar las causas de hipercolesterolemia con genotipos negativos. Para el cálculo de este score se evalúa la presencia de polimorfismos de nucleótido único (SNPs) en genes seleccionados involucrados con el metabolismo de LDL (14).
El estudio del perfil lipídico actual en el laboratorio clínico para diagnóstico de dislipemias y clasificación del riesgo cardiovascular consiste en un estudio básico estándar, que incluye colesterol total, C-HDL, C-LDL y triglicéridos y un estudio expandido con la medición de apoB -como el mejor marcador de riesgo de ECV- y la lipoproteína "a" [Lp(a)] que es una lipoproteína aterotrombótica y pro-inflamatoria cuyo nivel está determinado genéticamente y actualmente se impone su medida en todos los individuos para una correcta clasificación de riesgo y establecer conductas terapéuticas cuando se encuentra elevada. La particularidad es que Lp(a) contiene LDL en su estructura y constituye una interferencia cuando se mide o calcula C-LDL, es por eso que, ante la sospecha de HF la medida de Lp(a) elevada explica un resultado de C-LDL incrementado que no es propio de la lipoproteína LDL. Algunos autores propusieron la asociación concomitante de HF y Lp(a) elevada y el tema es aún controvertido (15).
En base a lo expuesto surge la hipótesis de que existe gran variabilidad en el desarrollo acelerado de aterosclerosis entre los individuos con hipercolesterolemias severas, como la HF, lo cual podría deberse no sólo al nivel elevado de colesterol acumulativo sino también a presencia de variantes patogénicas y/o poligénicas.
Objeto de Estudio
- Abordar al subgrupo de pacientes con HS con testeo genético, del Estudio Da Vinci, llevado a cabo en gran parte en la Ciudad de Mar del Plata, Partido de Gral. Pueyrredón.
Objetivos general:
- Evaluar las características clínicas, bioquímicas y genéticas de los pacientes HS
- Asociar la relación entre el fenotipo con elevación extrema de C-LDL y la presencia de variantes genéticas en los genes vinculados
- Detectar HF en portadores de variantes monogénicas,
- Explicar las causas de la fisiopatología de la HS mediante las variantes poligénicas.
- Determinar la concentración de Lp(a) y evaluar si pacientes con HF definida presenta mayores niveles
FUNDAMENTACIÓN TEÓRICA
El riesgo de la ECV se relaciona directamente con la exposición crónica y acumulativa de los niveles elevados de las LDL. La HS se define cuando el valor del C-LDL es mayor a 190 mg/dL. La HF es un desorden monogénico autosómico dominante, que causa aterosclerosis prematura severa y elevada mortalidad. Según diferentes registros poblacionales la prevalencia se estima en 1/200-1/300 para heterocigotas y 1/600.000 para las formas homocigotas.
La problemática mundial actual es el subdiagnóstico y en consecuencia, el subtratamiento de la HF y la imposibilidad de realizar el rastreo de familiares directos a fin de detectar otros portadores y comenzar con el inicio temprano del tratamiento. En nuestro país, el único estudio de detección de HS y de prevalencia de la HF se inició en la Ciudad de Mar del Plata en el año 2014. El avance y profundización en las características clínicas, bioquímicas y genéticas de esta población objeto del estudio, aportará al conocimiento de las particularidades de nuestra población, esclarecerá la la correlación entre el fenotipo, el diagnóstico clínico-bioquímico y el genético y la controvertida asociación de Lp(a) con HF.
MATERIAL Y MÉTODOS
Los pacientes mayores de 18 años, dentro del Registro DA VINCI para la detección y caracterización de pacientes con Hipercolesterolemia Familiar en Argentina, fueron seleccionados de dos bases de datos; una de 51.253 sujetos proporcionada por la Secretaría de Salud del distrito de General Pueyrredón y la otra proveniente del Departamento de Lípidos y Aterosclerosis del Instituto de Clínica Médica de Mar del Plata. Se convocaron 500 pacientes en un período comprendido entre Abril del 2014 y Febrero del 2022. El criterio de inclusión para participar en este estudio fue un nivel de colesterol sérico total >300 mg/dL y/o un C-LDL >190 mg/dL; como criterios de exclusión se consideraron la presencia de enfermedad renal avanzada, colestasis hepática, hipotiroidismo no tratado, diabetes mal controlada, embarazo y fármacos que alteran los niveles de colesterol. A un total de 122 sujetos que presentaron score de DUTCH > 6 se les realizó estudio genético, así como una completa evaluación clínica y bioquímica y constituyen el objeto del presente estudio. Se obtuvo el consentimiento informado de todos los pacientes mediante un protocolo y formulario de consentimiento aprobado por el Comité de Ética de la Universidad de Buenos Aires (Res CD 4705/14).
Adicionalmente al estudio bioquímico y genético, se obtuvo una historia extensa con un enfoque especial en ECV; se relevaron datos antropométricos, se realizó un examen clínico completo y finalmente se solicitaron datos sobre antecedentes personales y familiares de ECV. Dentro de los antecedentes cardiovasculares (CV) se recolectaron antecedentes de angina de pecho, infarto de miocardio, angioplastia coronaria, cirugía revascularización coronaria, accidente cerebrovascular isquémico y/o enfermedad vascular periférica o carotídea, hipertensión arterial, diabetes mellitus, tabaquismo y tratamiento hipolipemiante actual. Se realizó una cuidadosa exploración física con especial cuidado en detectar la presencia de xantomas tendinosos y/o arco corneal. Se obtuvieron y examinaron los registros médicos de los casos índice y, cuando estaban disponibles, de sus familiares de primer grado (padres, hijos y hermanos). La ECV prematura se definió como la presencia de la misma antes de los 55 años en los casos índice masculinos y familiares, y antes de los 65 años en los casos índice femeninos y familiares. Con los antecedentes familiares y personales de cada paciente, los signos clínicos y el valor más alto de C-LDL observado se calculó el score de DUTCH (Tabla 1). Aquellos pacientes que estaban recibiendo tratamiento hipolipemiante, y cuando no se disponía de datos de C-LDL sin medicación, se aplicó una corrección de C-LDL en función de la dosis y la potencia de la estatina utilizada (16). A todos estos sujetos se les extrajo sangre sin requerir ayuno y las muestras se enviaron al Laboratorio de Lípidos y Aterosclerosis de la Universidad de Buenos Aires para su análisis bioquímico.
-Estudios Bioquímicos: El CT, los TG, el C-HDL, el C-LDL directo, la creatinina y la fosfatasa alcalina se midieron en muestras de suero utilizando métodos enzimáticos estandarizados con reactivos de Roche Diagnostics, Mannheim/Alemania, utilizando un autoanalizador Cobas C-501. Los valores medios de los coeficientes de variación (C.V.) para estos parámetros fueron < 2,3 % para los CVa intraensayo y < 3,0 % para los C.V. interensayo. Los niveles séricos de apolipoproteína B (apoB) se determinaron utilizando un ensayo inmunoturbidimétrico de Roche en el mismo analizador automatizado, con C.V. intraensayo e interensayo de <2,5 %. La Lp(a), se midió por inmunoturbidimetría automatizada en sistemas COBAS INTEGRA (ROCHE), con anticuerpos policlonales anti-Lp(a) y material de referencia -Preciset Lp(a) Gen2- compuesto por cinco calibradores basados en pool de plasmas humanos estabilizados y liofilizados, con trazabilidad a SRM2B, IFCC/OMS, independientemente del tamaño de apo(a). Los resultados de Lp(a) se expresaron en nmol/L. La TSH se midió por quimioluminiscencia (DPC, Immulite, Los Ángeles, CA, EE. UU.) con C.V. intra e interensayo <3,5 %.
-Estudios Genéticos: Se extrajo el ADN genómico de sangre completa de los 122 pacientes, mediante la técnica Salting Out y se prepararon según el protocolo nacional de transporte muestras biológicas para su envío a dos Laboratorios en el exterior donde se llevaron a cabo las secuenciaciones genéticas: Boston Heart Lab, Boston Massachusetts-USA y posteriormente al Instituto Nacional de Saúde Doutor Ricardo Jorge, Lisboa, Portugal. La secuenciación de ADN se llevó a cabo en un instrumento Illumina MiSeq Dx utilizando 2 x 150 lecturas finales emparejadas y se realizó en los siguientes loci de genes (exones e intrones) en relación al aumento de los niveles séricos de C-LDL: LDLR, APOB, PCSK9, LDLRAP1 y APOE.La mediana de la profundidad de lectura fue de 600X, con una sensibilidad y especificidad del 100 % para SNV y una sensibilidad del 100 % y una especificidad del 91 % para Indel. Los archivos FASTQ se procesaron mediante un flujo de trabajo personalizado en CLC Biomedical Genomics Workbench (v3.2, Qiagen). Los archivos VCF que contenían todas las variantes identificadas en las regiones seleccionadas se anotaron mediante Ingenuity Variant Analysis (Qiagen) con una cascada de filtrado personalizada. Las variantes se clasificaron según el Colegio Americano de Genética Médica y Genómica (ACMG) (17). Además, la evaluación de las CNV y las variantes estructurales grandes se realizó utilizando herramientas integradas de Biomedical Genomics Workbench y VarSeq CNV Analysis de Golden Helix. Las clasificaciones de variantes se basaron en las pautas de ACMG.
Sumado al análisis de variantes monogénicas, se evaluaron 10 polimorfismos de nucleótido único (SNPs): rs6544713 en ABCG8, rs515135 en APOB, rs12740374 en CELSR2, rs3846663 en HMGCR, rs2650000 en HNF1, rs6511720 en R-LDL, rs6102059 en MAFB, rs10401969 en NCAN, rs11206510 en PCSK9, and rs1501908 en TIMD4, que constituyeron la determinación de un score de riesgo poligénico (GRS) desarrollado para evaluar la contribución poligénica al aumento de C-LDL (18). Para el GRS se utilizó un punto de corte correspondiente al percentilo 90th de 1.96, con un valor máximo de 2,42.
-Análisis estadístico: se analizó la frecuencia y distribución de las variables a nivel global, por sexo y por score de DUTCH . Los datos categóricos se presentan en forma de porcentajes y los continuos utilizando la media para la tendencia central y el desvío estándar para dispersión en caso de distribución normal. En caso de datos continuos de distribución distinta de la normal se utilizaron como medidas de resumen la mediana para la tendencia central y los percentilos para la dispersión.
Las variables categóricas se analizaron por el método de Chi cuadrado con corrección de Fisher cuando fue necesario. Las variables numéricas con distribución normal se analizaron con el test t de Student, y las numéricas con distribución asimétrica con el test de Wilcoxon. Para todos los análisis estadísticos se consideró significativo un valor de p<0,05 a dos colas.
El análisis estadístico se realizó utilizando el Software estadístico Stata versión 11.0 (StataCorp. Stata Statistical Software: Release 11. College Station, TX: StataCorp LP; 2009).
Tabla 1- Criterios de la Red de Clínicas de Lípidos holandesas. Estimación del Score de DUTCH para el diagnóstico clínico de hipercolesterolemia familiar
Resultados
Se analizaron 122 pacientes con HS siendo la edad media de 50,6 ±10 y el 67,75% de sexo femenino. La mediana del score de DUTCH fue de 7; rango intercuartilo (RIC) 3-17. Las características clínicas de los sujetos estudiados se presentan en la Figura 1 y las características bioquímicas en la Tabla 2. El 41,6% (n= 50) de los participantes estaban tratados con estatinas, por lo cual explica que la media de C-LDL fue menor de 190 mg/dl. En la Tabla 3 se observan las características clínicas y bioquímicas evaluadas según sexo, observando que los hombres presentaron mayores valores de CT, C-LDL y menores de C-HDL (p < 0,05), estos resultados se aprecian ilustrados en la Figura 2.

Figura 1. Características clínicas de la población.
Tabla 2. Parámetros lipídicos en la muestra total.
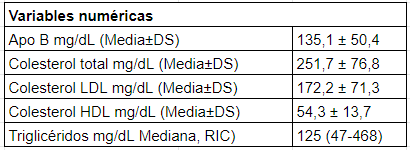
RIC: rango intercuartilo
Tabla 3. Características clínicas ybioquímicas de la población x sexo
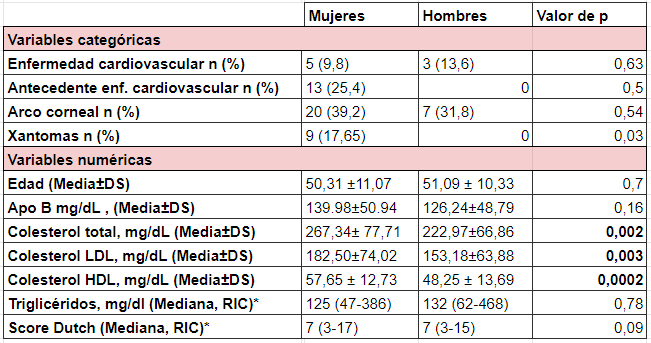
RIC: rango intercuartilo. Se aplicó Chi cuadrado con corrección de Fisher en las variables categóricas y en las variables numéricas T-test o test de Wilcoxon (*) según distribución.
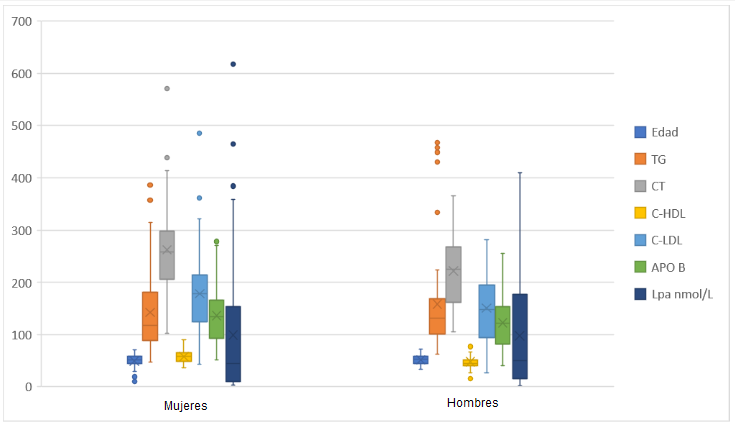
Figura 2. Representación gráfica de la variables cuantitativas -edad y parámetros lipídicos- analizadas por sexo.
En la Tabla 4 se observan las características bioquímicas y clínicas de los sujetos subdivididos según score de DUTCH menor de 9 y mayor o igual a 9. Según lo esperado, estos últimos demostraron claramente valores más altos de colesterol total, C-LDL y apoB y mayor proporción de individuos con xantomas. Por otro lado, el análisis univariado de los posibles predictores de hipercolesterolemia evidenció que la presencia de xantomas y de arco corneal son fuertes predictores de presentar score de DUTCH mayor o igual a 9 (Tabla 5).
Tabla 4. Parámetros bioquímicos estratificados por Score de Dutch ≥9
|
|
DUTCH < 9 |
DUTCH ≥ 9 |
Valor de p |
|
ApoB mg/dL (Media±DS) |
126,10±45,9 |
156,88±54,72 |
0,0024 |
|
Colesterol total mg/dL (Media±DS) |
267,34±66 |
290,82±90 |
0,0003 |
|
Colesterol LDL mg/dL (Media±DS) |
156,6±60,21 |
210,2±85,87 |
0,0002 |
|
Colesterol HDL mg/dL (Media±DS) |
55,23±14,62 |
51,32±11,03 |
0,1331 |
|
Triglicéridos mg/dL (Mediana, RIC)* |
128 (47-468) |
128 (48-314) |
0,602 |
RIC: rango intercuartilo. Las variables numéricas con distribución normal se analizaron con T-test y las que presentaron con distribución no normal (*) con test de Wilcoxon.
Tabla 5. Análisis univariado de predictores para la presencia de Score de Dutch≥9. OR: odds ratio.
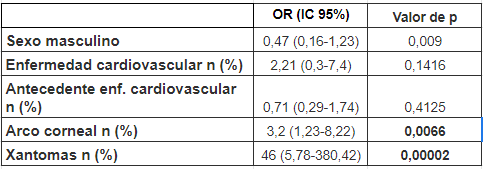
Las variables categóricas fueron analizadas con Chi cuadrado con corrección de Fisher
En cuanto al análisis genético, se encontraron variantes en genes involucrados en el 55% de los participantes siendo en el de estos 79% una variante monogénica, 14,4% causas poligénicas y solo 4,4% presentó variantes monogénicas y poligénicas (ver Figura 3). Comparando los pacientes con presencia de variantes versus ausencia, se encontró como significativo niveles más elevados de CT y C-LDL en aquellos portadores de variantes (p<0,05), no encontrándose diferencias en las características clínicas. En cuanto a los niveles de Lp(a) presentaron valores mayores al valor de corte de 125 mmol el 24% de los participantes con mutaciones versus el 33% de los que no tenían mutaciones (p=0,023).

Figura 3. Flujograma de los resultados del análisis genético.
P: variante patogénica (pathogenic); LP: variante probablemente patogénica (Likely pathogenic);
VUS: variante significado incierto (variant of uncertain significance); LDLR: gen que codifica para el receptor de LDL
APOB: gen que codifica para la apolipoproteína B; PCSK9: gen que codifica para la pro-proteína substilisina kexina tipo-9
DISCUSIÓN
El presente trabajo describe en forma detallada y precisa, por primera vez en nuestro país, las características clínicas, bioquímicas y genéticas de una población con HS del Municipio de General Pueyrredón. Este estudio permitió analizar, por primera vez en Argentina, un grupo de 122 individuos en una determinada localidad de la Provincia de Buenos Aires, que presentaron una alteración en su perfil lipídico-lipoproteico plasmático específico como es la HS. La detección bioquímica de un valor de C-LDL > 190 mg/dL, luego de descartar las causas secundarias, condujo a la caracterización clínica de cada uno de estos individuos y finalmente se realizó un diagnóstico genético en centros de primer nivel, especializados en trastornos dislipidémicos hereditarios, en Portugal y EE. UU, con el fin de detectar pacientes portadores de hipercolesterolemia familiar e implementar un programa de registro del HF .
En el año 2014 comenzamos en el Municipio de General Pueyrredón, el primer estudio de detección de HF en Argentina denominado DA VINCI. A partir de esa fecha se recolectaron resultados provenientes de dos bases de datos, una del Servicio de Salud del Municipio de General Pueyrredón y otra del Departamento de Lípidos y Aterosclerosis del Instituto de Clínica Médica de Mar del Plata. Luego de 7 años de estudio y seguimiento, se detectaron y analizaron 122 individuos que cumplimentaban con los criterios de inclusión propuestos para la realización del estudio genético. A todos estos pacientes se les realizó una exhaustiva evaluación clínica, con foco en su aparato cardiovascular y adicionalmente un profundo análisis bioquímico en relación a su dislipidemia, para finalmente concluir con estudios genéticos de última generación en busca de variantes y resultados que expliquen el fenotipo hallado en cada individuo. Se resalta que este análisis y evaluación detallada de este grupo de individuos se realizó por primera vez en Argentina y constituye un trabajo original en nuestra región. En el año 2017, nuestro grupo publicó resultados preliminares conjuntamente con otros países de Iberoamérica, que incluían un número reducido de pacientes de Argentina (19); el presente trabajo constituye un avance ya que logra profundizar el análisis y ampliar los datos clínicos y, bioquímicos y genéticos con el agregado fundamental de suscribirlos a una población específica como es la del Municipio de General Pueyrredón en la Argentina.
La población analizada presentóuna media de edad de 50 años y aproximadamente el 70% fueron mujeres; se encontraron diferencias estadísticamente significativas al comparar mujeres y hombres, a favor de las primeras en el valor de colesterol total y C-LDL. Esta diferencia se explicaría por el incremento significativo de colesterol en la postmenopausia, debido al descenso de estrógenos y disminución de los receptores de LDL, teniendo en cuenta que 62 % del grupo de mujeres presentaban más de 50 años (20).
En referencia a los predictores de score DUTCH ≥ 9 -considerado un puntaje de diagnóstico definitivo, se hallaron como variables con significancia estadística a la presencia de arco corneal y xantomas tendinosos, junto a los parámetros bioquímicos de colesterol total, apoB y C-LDL; esto va en consonancia con el valor de cada uno de estos predictores en el diagnóstico clínico de la HF según los criterios DUTCH (10). El valor medio de C-LDL hallado fue de 170 mg/dL; si bien este valor es inferior a lo que se considera HS, se debería al grupo de individuos que se encontraban bajo tratamiento hipolipemiante al momento del enrolamiento. De hecho, un 40% de los pacientes se encontraba recibiendo tratamiento al momento del contacto y evaluación, donde predominaba la utilización de estatinas en baja dosis (98%), alcanzando el valor de C-LDL < 100 mg/dL en solo el 8% de los casos. Al realizar el ajuste a valores basales (sin tratamiento farmacológico) de C-LDL, aplicando la fórmula correctiva propuesta por Haralambos et.al. (16), basada en un factor según tipo y dosis de fármaco que recibe el paciente, este valor de C-LDL se sitúa en una media y desvío de 209 mg/dL ± 18 mg/dL. Cabe destacar que los pacientes convocados por cumplir con el criterio de inclusión, un porcentaje relevante de individuos que participaron del estudio, presentó valores inferiores a 190 mg/dL, aunque lejos de las metas requeridas para disminuir su riesgo, lo cual, sumado a los que no recibieron ningún tratamiento hipolipemiante, confirma la problemática mundial del subtratamiento y subdiagnóstico de los pacientes con HS y/o HF (5).
En referencia a los estudios genéticos realizados, se han analizado las variantes monogénicas clásicas relacionadas a la HF (LDLR, APOB, PCSK9, LDLRAP1 y APOE) así como un panel de 10 SNPs que determinaron un score de riesgo poligénico (GRS) desarrollado para analizar la contribución poligénica al aumento de los niveles de C-LDL. De los 122 individuos estudiados, 55% presentaron un test genético positivo y dentro de esta proporción el 80% mostraron una variante monogénica, un 16% un patrón poligénico y un 4% compartieron una variante monogénica asociada a un patrón poligénico. Cabe destacar que de los 53 pacientes que evidenciaron una variante monogénica, 45% fueron patogénicas o probablemente patogénicas y un 41% clasificadas como variantes de significado incierto (VUS).
Diferentes estudios y publicaciones de otras latitudes, que incluyeron análisis similares a los del presente trabajo, muestran resultados comparables a los hallazgos mencionados; un 50% de los individuos con niveles de C-LDL > 190 mg/dL poseen un estudio genético positivo, donde predomina la presencia de una variante monogénica en los genes característicos de la HF, especialmente LDLR, completando este porcentaje con un GRS positivo que explican las causas de la hipercolesterolemia (21, 22).
Es interesante y recientemente descripto que, entre los pacientes con HS con estudios genéticos negativos, se incluye un grupo de individuos con elevados valores de Lp(a); esta lipoproteína aterogénica tiene en su composición un porcentaje de colesterol que contribuye al valor final de colesterol total y al de C-LDL. Por lo tanto, cuando Lp(a) está elevada, el aporte de su colesterol a la medida del C-LDL, explicaríael fenotipo hallado. De hecho, los individuos con Lp(a) incrementada, presentaron una menor proporción (p<0.023) de presencia de variantes genéticas asociadas a HF, con lo cual se deduce que el incremento de C-LDL de estos pacientes es a expensas de la contribución del colesterol de la Lp(a) (23, 24).
La relación causal entre los elevados niveles de C-LDL y la enfermedad cardiovascular aterosclerótica está demostrada por todos los niveles de evidencia disponible en la actualidad (1). Más allá de esto, se conoce que la carga aterogénica que padece una persona, es directamente proporcional a los niveles de C-LDL multiplicado por el tiempo de exposición a este factor de riesgo lipídico (25). Niveles de C-LDL por encima de 190 mg/dL se consideran extremadamente elevados, denominándose a esta condición, cómo HS. Luego de descartar las causas secundarias de este trastorno lipídico, es imperativo el plantear el diagnóstico de HF como primera alternativa, ya que su detección implica una serie de conductas obligadas, como la evaluación cardiovascular exhaustiva, el rastreo de familiares directos y la instauración de un tratamiento intensivo a la brevedad (26). Si bien los estudios genéticos son cada vez más accesibles, y en la medicina personalizada suele evaluarse, no es requisito imprescindible para arribar a un diagnóstico certero, para llevar a cabo cascada familiar a partir de un caso índice ni para indicar el tratamiento correspondiente lo más temprano posible (1).
De la población estudiada y analizada, se desprende que el 46% presentó un test genético positivo monogénico (patogénico, probablemente patogénico o de significado incierto), pudiendo afirmar que estos pacientes son portadores de HF, con las implicancias en cuanto a la necesidad de tratamiento intensivo y la imperiosa detección de nuevos casos en el resto de los familiares. Si bien todas las HS deben ser correctamente tratadas, ya sea si son secundarias a otras patologías, se deban a causas poligénicas, o sean HF por causas monogénicas, hay evidencias claras de que a un mismo nivel de C-LDL circulante, los portadores de variantes monogénicas presentan mayor riesgo de producir eventos cardiovasculares, dado que han mantenido niveles mayores de C-LDL desde sus primeros años de vida (1, 11).
CONCLUSIÓN
Como conclusión podemos afirmar que hemos detectado y analizado detalladamente, en tres diferentes esferas -clínica, bioquímica y genética- a un grupo de 122 individuos provenientes del Municipio de General Pueyrredón, en un período de tiempo de 7 años, con diagnóstico primario HS, siendo 56 portadores de HF. La mayoría de estos pacientes no estaba siendo tratado acorde a las guías y recomendaciones establecidas y presentaban en un 55% un resultado genético positivo, siendo las variantes monogénicas relacionadas a la HF las más frecuentemente detectadas (80%). Los hallazgos clínicos, como la presencia de xantomas tendinosos y la detección del arco corneal precoz, así como las variables bioquímicas (colesterol total, C-LDL y apoB) fueron predictores estadísticamente significativos de un score de DUTCH ≥9 (diagnóstico definitivo de HF) así como de la presencia de un resultado positivo en el análisis genético de esta población.
Bibliografía
- Santos RD, Gidding SS, Hegele RA, Cuchel MA, Barter PJ, Watts GF, et al. International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016;4(10):850–61.
- Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459–72
- Goldstein JL, Hobbs HH, Brown MS. The metabolic and molecular basis of inherited disease. Scriver CT, Beaudet AL, Sly WS, Valle D, editors. New York: McGraw–Hill; 1995.
- Bell DA, Watts GF. Progress in the care of familial hypercholesterolaemia: 2016. Med J Aust. 2016;205(5):232–6.
- Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478–90a.
- Corral P, Geller AS, Polisecki EY, Lopez GI, Bañares VG, Cacciagiu L, et al. Unusual genetic variants associated with hypercholesterolemia in Argentina. Atherosclerosis. 2018;277:256–61.
- Arca M. Old challenges and new opportunities in the clinical management of heterozygous familial hypercholesterolemia (HeFH): The promises of PCSK9 inhibitors. 2017;256:134–45.
- Goldstein JL, Brown MS. A century of cholesterol and coronaries: From plaques to genes to statins. 2015;161(1):161–72.
- Futema M, Plagnol V, Whittall RA, Neil HAW, Simon Broome Register Group, Humphries SE, et al. Use of targeted exome sequencing as a diagnostic tool for Familial Hypercholesterolaemia. J Med Genet. 2012;49(10):644–9.
- Programme WHG. Familial hypercholesterolaemia (FH) : report of a second WHO consultation, Geneva, 4 September 1998. World Health Organization; 1999.
- Khera AV, Won H-H, Peloso GM, Lawson KS, Bartz TM, Deng X, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67(22):2578–89.
- Corral P, Bañares V, Sáenz B, Zago V, Sarobe A, López G, et al. Phenotype of definite familial hypercholesterolemia with negative genetic study in Argentina. Arch Cardiol Mex. 2020;90(2):130–6.
- Medeiros AM, Bourbon M. Polygenic contribution for familial hypercholesterolemia (FH). Curr Opin Lipidol. 2021;32(6):392–5.
- Futema M, Shah S, Cooper JA, Li K, Whittall RA, Sharifi M, et al. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries. Clin Chem. 2015;61(1):231–8.
- Sjouke B, Yahya R, Tanck MWT, Defesche JC, de Graaf J, Wiegman A, et al. Plasma lipoprotein(a) levels in patients with homozygous autosomal dominant hypercholesterolemia. J Clin Lipidol. 2017;11(2):507–14.
- Haralambos K, Whatley SD, Edwards R, Gingell R, Townsend D, Ashfield-Watt P, et al. Clinical experience of scoring criteria for Familial Hypercholesterolaemia (FH) genetic testing in Wales. 2015;240(1):190–6.
- Richards S, Aziz N, Bale S, Bick D, Das S. ACMG laboratory quality assurance committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology, Genet. Genet Med. 2015;17:405–e424.
- Wang JS, Dron MR, Ban JF, Robinson AD. Polygenic versus monogenic causes of hypercholesterolemia ascertained clinically, Arterioscler. Arterioscler Thromb Vasc Biol. 2016;36:2439–e2445.
- Santos RD, Bourbon M, Alonso R, Cuevas A, Vasques-Cardenas NA, Pereira AC, et al. Clinical and molecular aspects of familial hypercholesterolemia in Ibero-American countries. J Clin Lipidol. 2017;11(1):160-166.
- Berg G, Mesch V, Boero L, Sayegh F, Prada M, Royer M, et al. Lipid and lipoprotein profile in menopausal transition. Effects of hormones, age and fat distribution. Horm Metab Res. 2004;36(4):215–220.
- Jacob E, Hegele RA. Monogenic Versus Polygenic Forms of Hypercholesterolemia and Cardiovascular Risk: Are There Any Differences?. Curr Atheroscler Rep. 2022;24(6):419–426.
- D’Erasmo L, Minicocci I, Di Costanzo A, Pigna G, Commodari D, Ceci F, et al. Clinical Implications of Monogenic Versus Polygenic Hypercholesterolemia: Long-Term Response to Treatment, Coronary Atherosclerosis Burden, and Cardiovascular Events. J Am Heart Assoc. 2021;10(9):e018932.
- Langsted A, Nordestgaard BG. Lipoprotein(a) as Part of the Diagnosis of Clinical Familial Hypercholesterolemia. Curr Atheroscler Rep. 2022;24(4):289-296.
- Yeang C, Willeit P, Tsimikas S. The interconnection between lipoprotein(a), lipoprotein(a) cholesterol and true LDL-cholesterol in the diagnosis of familial hypercholesterolemia. Curr Opin Lipidol. 2020;31(6):305–312.
- Zambon A, Mello E Silva A, Farnier M. The burden of cholesterol accumulation through the lifespan: why pharmacological intervention should start earlier to go further?. Eur Heart J Cardiovasc Pharmacother. 2021;7(5):435–441.
- Tokgozoglu L, Kayikcioglu M. Familial Hypercholesterolemia: Global Burden and Approaches. Curr Cardiol Rep. 2021;23(10):151.